| Heterogeneous Catalysis | |
| Energetic Materials | |
| Bio-active Molecules |
|
On The Excited Electronic State Decomposition of Nitramine Energetic Materials and Their Model Systems
| |
|
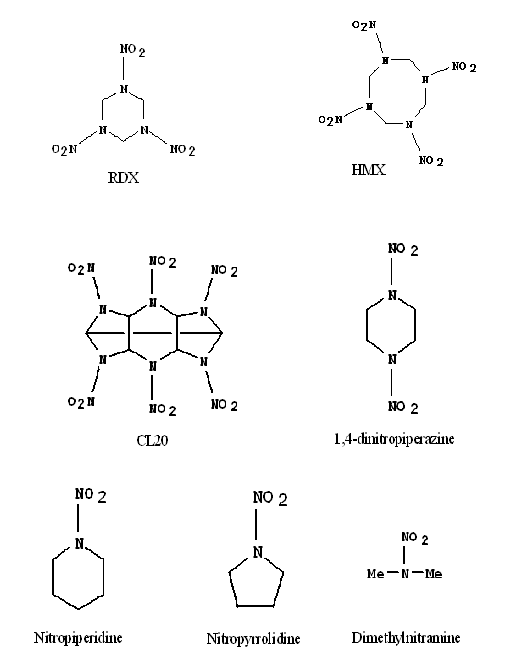
In order to elucidate the difference between nitramine energetic materials, such as RDX (1,3,5-trinitro-1,3,5-triazacyclohexane), HMX (1,3,5,7-tetranitro-1,3,5,7-tetraazacyclooctane), and CL-20 (2,4,6,8,10,12-hexanitro-2,4,6,8,10,12-hexaazaisowurtzitane), and their non-energetic model systems, including 1,4-dinitropiperazine, nitropiperidine, nitropyrrolidine and dimethylnitramine, both nanosecond mass resolved excitation spectroscopy (MRES) and femtosecond pump-probe spectroscopy in the UV spectral region have been employed to investigate the mechanisms and dynamics of the excited electronic state photodissociation of these materials. The NO molecule is an initial decomposition product of all systems. The NO molecule from the decomposition of energetic materials displays cold rotational and hot vibrational spectral structures. Conversely, the NO molecule from the decomposition of model systems shows relatively hot rotational and cold vibrational spectra. Another potential initial product of nitramine excited electronic state dissociation could be OH, generated along with NO, perhaps from an HONO intermediate species. The OH radical is not observed in fluorescence even though its transition intensity is calculated to be 1.5 times that found for NO per radical generated. The HONO intermediate is thereby found not to be an important pathway for the excited electronic state decomposition of these cyclic nitramines. Parent ions of dimethylnitramine and nitropyrrolidine are observed in femtosecond experiments. All the other molecules generate NO as a decomposition product even in the femtosecond time regime. The dynamics of the formation of the NO product is faster than 180 fs, which is equivalent to the time duration of our laser pulse. | |
|
Chemical Structures of Nitramine Energetic Materials and Model Systems | |
|
| |
|
1-Color MRES of The A2Σ (v'=0) ← X2Π (v''=0,1,2,3) Transitions of NO from CL-20 | |

| |
|
1-Color MRES of The A2Σ (v'=0) ← X2Π (v''=0) Transition of NO from Nitramine Model Systems | |

| |
|
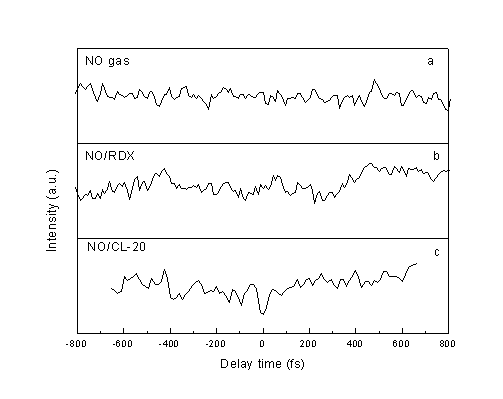
fs Pump-Probe Transients for The NO Molecule from NO Gas (a), and The Photodissociation of RDX (b) and CL-20 (c) at 226 nm | |
| |
|
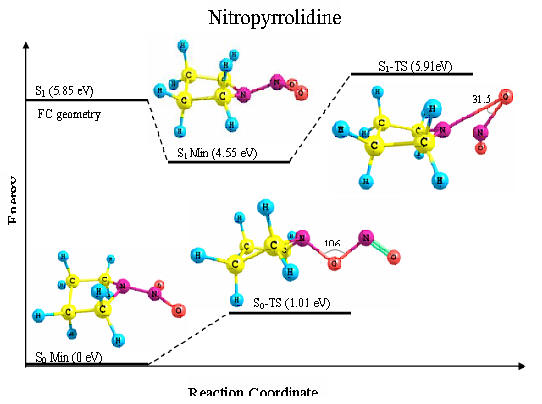
Potential energies and structures of the transition states of nitropyrrolidine in its ground and first excited electronic states. Energies are in eV | |
|
|